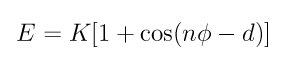
Syntax:
fix ID group-ID restrain Kstart Kstop keyword value(s)
dihedral value = atom1 atom2 atom3 atom4 target
atom1,atom2,atom3,atom4 = IDs of 4 atoms in restrained dihedral
target = target value for specified dihedral angle (degrees)
Examples:
fix holdem all restrain 2000.0 2000.0 dihedral 1 2 3 4 120.0 fix texas_holdem all restrain 0.0 2000.0 dihedral 1 2 3 4 120.0 1 2 3 5 -120.0 1 2 3 6 0.0
Description:
Restrain the motion of the specified atoms by making them part of a bond or angle or dihedral interaction whose strength can vary over time during a simulation. This is functionally equivalent to creating a bond or angle or dihedral for the atoms in a data file, as specified by the read_data command, albeit with a time-varying pre-factor coefficient. For the purpose of forcefield parameter-fitting or mapping a molecular potential energy surface, this fix reduces the hassle and risk associated with modifying data files. In other words, use this fix to temporarily force a molecule to adopt a particular conformation. To form a permanent bond or angle or dihedral, modify the data file.
The first example above applies a restraint to hold the dihedral angle formed by atoms 1, 2, 3, and 4 near 120 degrees using a constant restraint coefficient. The second example applies similar restraints to multiple dihedral angles using a restraint coefficient that increases from 0.0 to 2000.0 over the course of the run.
IMPORTANT NOTE: Adding a force to atoms implies a change in their potential energy as they move due to the applied force field. For dynamics via the run command, this energy can be added to the system's potential energy for thermodynamic output (see below). For energy minimization via the minimize command, this energy must be added to the system's potential energy to formulate a self-consistent minimization problem (see below).
In order for a restraint to be effective, the restraint force must typically be significantly larger than the forces associated with conventional forcefield terms. If the restraint is applied during a dynamics run (as opposed to during an energy minimization), a large restraint coefficient can significantly reduce the stable timestep size, especially if the atoms are initially far from the preferred conformation. You may need to experiment to determine what value of K works best for a given application.
For the case of finding a minimum energy structure for a single molecule with particular restratins (e.g. for fitting forcefield parameters or constructing a potential energy surface), commands such as the following might be useful:
# minimize molecule energy with restraints velocity all create 600.0 8675309 mom yes rot yes dist gaussian fix NVE all nve fix TFIX all langevin 600.0 0.0 100 24601 fix REST all restrain 0.0 5000.0 dihedral 2 1 3 8 $angle1 3 1 2 9 $angle2 fix_modify REST energy yes run 10000 fix TFIX all langevin 0.0 0.0 100 24601 fix REST all restrain 5000.0 5000.0 dihedral 2 1 3 8 $angle1 3 1 2 9 $angle2 fix_modify REST energy yes run 10000 # sanity check for convergence minimize 1e-6 1e-9 1000 100000 # report unrestrained energies unfix REST run 0
The dihedral keyword applies a dihedral restraint to the specified atoms using a simplified form of the function used in dihedral_style charmm. Specifically, the potential associated with the restraint is
with the following coefficients:
Restart, fix_modify, output, run start/stop, minimize info:
No information about this fix is written to binary restart files.
The fix_modify energy option is supported by this fix to add the potential energy associated with this fix to the system's potential energy as part of thermodynamic output.
IMPORTANT NOTE: If you want the fictitious potential energy associated with the added forces to be included in the total potential energy of the system (the quantity being minimized), you MUST enable the fix_modify energy option for this fix.
This fix computes a global scalar, which can be accessed by various output commands. The scalar is the potential energy discussed above. The scalar value calculated by this fix is "extensive".
No parameter of this fix can be used with the start/stop keywords of the run command.
Restrictions:
The group-ID specified by this fix is ignored.
Currently, only dihedral restraints are allowed, but modification of the code to allow angle and bond restraints would be straightforward.
Related commands: none
Default: none