| IMIX | = | type of mixing |
| AMIX | = | linear mixing parameter |
| AMIN | = | minimal mixing parameter |
| BMIX | = | cutoff wave vector for Kerker mixing scheme |
| AMIX_MAG | = | linear mixing parameter for magnetization |
| BMIX_MAG | = | cutoff wave vector for Kerker mixing scheme for mag. |
| WC | = | weight factor for each step in Broyden mixing scheme |
| INIMIX | = | type of initial mixing in Broyden mixing scheme |
| MIXPRE | = | type of preconditioning in Broyden mixing scheme |
| MAXMIX | = | maximum number steps stored in Broyden mixer |
Default (please rely on these defaults)
| US-PP | PAW | ||
| IMIX | = | 4 | 4 |
| AMIX | = | 0.8 | 0.4 |
| BMIX | = | 1.0 | 1.0 |
| WC | = | 1000. | 1000. |
| INIMIX | = | 1 | 1 |
| MIXPRE | = | 1 | 1 |
| MAXMIX | = | -45 | -45 |
MAXMIX is only available in VASP.4.4 and newer versions, and it is strongly recommended to use this option for molecular dynamics and relaxations.
With the default setting, a Pulay mixer[26]
with an initial approximation for the charge dielectric function according to
Kerker, Ref. [41]
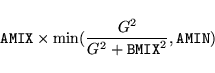 |
(11) |
| US-PP | PAW | ||
| AMIX | = | 0.4 | 0.4 |
| AMIN | = | 0.1 | 0.1 |
| BMIX | = | 1.0 | 1.0 |
| AMIX_MAG | = | 1.6 | 1.6 |
| BMIX_MAG | = | 1.0 | 1.0 |
The above setting is equivalent to an (initial) spin enhancement
factor of 4, which is usually a reasonable approximation.
There are only a few other parameter combinitions
which can be tried, if convergence turns out to be very slow. In particular,
for slabs, magnetic systems and insulating systems
(e.g. molecules and clusters),
an initial ``linear mixing'' can result in faster convergence than
the Kerker model function.
One can therefore try to use the following setting
| AMIX | = | 0.2 |
| BMIX | = | 0.0001 ! almost zero, but 0 will crash some versions |
| AMIX_MAG | = | 0.8 |
| BMIX_MAG | = | 0.0001 ! almost zero, but 0 will crash some versions |
In VASP.4.x the eigenvalue spectrum of the charge dielectric matrix is calculated and written to the OUTCAR file at each electronic step. This allows a rather easy optimization of the mixing parameters, if required. Search in the OUTCAR file for
eigenvalues of (default mixing * dielectric matrix)The parameters for the mixing are optimal if the mean eigenvalue is 1, and if the width of the eigenvalue spectrum is minimal. For an initial linear mixing ( BMIX
One important option which might help to reduce the number of iterations for MD's and ionic relaxations is the option MAXMIX, which is only available in VASP.4.4. MAXMIX specifies the maximum number of vectors stored in the Broyden/Pulay mixer, in other words it corresponds to the maximal rank of the approximation of the charge dielectric function build up by the mixer. MAXMIX can be either negative or positive. If a negative value is specified for MAXMIX the mixer is reset after each ionic step or if the number of electronic steps exceeds abs( MAXMIX) (this is the default and similar to the behavior of VASP.4.3 and VASP.3.2). If MAXMIX is positive, the charge density mixer is only reset if the storage capabilities are exceeded. The reset is done ``smoothly'' by removing the five oldest vectors from the iteration history. Therefore, if MAXMIX is positive the approximation for the charge dielectric function which was obtained in previous ionic steps is ``reused'' in the current ionic step, and this in turn can reduce the number of electronic steps during relaxations and MD's. Especially for relaxations which start from a good ionic starting guess and for systems with a strong charge sloshing behavior the speedup can be significant. We found that for a 12 A long box containing 16 Fe atoms the number of electronic iterations decreased from 8 to 2-3 when MAXMIX was set to 40. For a carbon surface the number of iterations decreased from 7 to 3. At the same time the energy stability increased significantly. But be careful - this option increases the memory requirements for the mixer considerably, and thus the option is not recommended for systems were charge sloshing is negligible anyway (like bulk simple metals). The optimal setting for MAXMIX is usually around three times the number of electronic steps required in the first iteration. Too large values for MAXMIX might cause the code to crash (because linear dependencies between input vectors might develop).
Please go to the next section if you are not interested in
a more detailed dicussion of the flags that influence the mixer.
IMIX determines the type of mixing
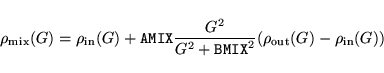 |
(12) |
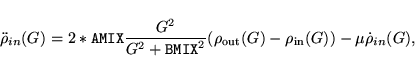
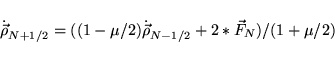
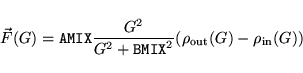
eigenvalues of (default mixing * dielectric matrix)in the OUTCAR file. The optimal parameters are then given by:
| AMIX | AMIX(as used in Pulay run)* smallest eigenvalue |
| AMIN= |
2*SQRT(smallest eigenvalue/ largest eigenvalue) |
The parameters WC, INIMIX and MIXPRE are meaningful only for the Broyden scheme:
WC determines the weight factors for each iteration
INIMIX determines the functional form of the initial mixing
matrix (i.e. ![]() for the Broyden scheme).
The initial mixing matrix might influence the convergence speed for complex
situations (especially surfaces and magnetic systems), nevertheless
INIMIX must not be changed from the default setting: anything
which can be done with INIMIX can also be done with AMIX and
BMIX, and
changing AMIX and BMIX is definitely preferable.
for the Broyden scheme).
The initial mixing matrix might influence the convergence speed for complex
situations (especially surfaces and magnetic systems), nevertheless
INIMIX must not be changed from the default setting: anything
which can be done with INIMIX can also be done with AMIX and
BMIX, and
changing AMIX and BMIX is definitely preferable.
Anyway, possible choices for INIMIX are:
MIXPRE determines the metric for the Broyden scheme
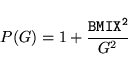 |
(13) |