For charged cells or for calculations of molecules
and surfaces with a large dipole moment, the energy converges
very slowly with respect to the size ![]() of the supercell.
Using methods discussed in Ref. [55,56]
VASP is able to correct for the leading errors, but
one should stress, that in many details,
we have taken a more general approach than that one
outlined in Ref. [55].
of the supercell.
Using methods discussed in Ref. [55,56]
VASP is able to correct for the leading errors, but
one should stress, that in many details,
we have taken a more general approach than that one
outlined in Ref. [55].
The following flags control the behavior of VASP.
NELECT determines the total number of electrons in the system
(see Sec. 6.34). For charged systems this
value has to be supplied by hand and a neutralizing
background charge is assumed by VASP. For these systems the energy
converges very slowly with respect to the size of the super cell.
The required first order energy correction is given by
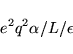
For systems with a net dipole moment the energy also converges
slowly with respect to the size of the super cell.
The dipole corrections (and quadrupole corrections for charged systems)
fall of like ![]() .
Both corrections (quadrupole only for charged
systems) will be calculated and added to the total energy
if the IDIPOL flag is set.
.
Both corrections (quadrupole only for charged
systems) will be calculated and added to the total energy
if the IDIPOL flag is set.
If set in the INCAR file monopole/dipole and quadrupole corrections will be calculated. There are four possible settings for IDIPOL
IDIPOL = 1-4For 1 to 3, the dipole moment will be calculated only into the direction of the first, second or third lattice vector. The corrections for the total energy are calculated as the energy difference between a monopole/dipole and quadrupole in the current supercell and the same dipole placed in a super cell with the corresponding lattice vector approaching infinity. This flag should be used for slab calculations.
For IDIPOL=4 the full dipole moment in all directions will be calculated, and the corrections to the total energy are calculated as the energy difference between a monopole/dipole/quadrupole in the current supercell and the same monopole/dipole/quadrupole placed in a vacuum, use this flag for calculations for isolated molecules.
DIPOL = center of cell (in direct, fractional coordinates)This tag determines as in VASP.3.2 the center of the net charge distribution. The dipol is defined as
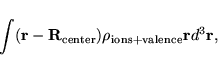 |
(15) |
This tag switches on the potential correction mode: Due to the periodic boundary conditions not only the total energy converges slowly with respect to the size of the supercell, but also the potential and the forces are ``wrong''. This effect can be counterbalanced by setting LDIPOL=.TRUE. in the INCAR file. In that case a linear and (in the case of a charged system) a quadratic electrostatic potential is added to the local potential correcting the errors introduced by the periodic boundary conditions. This is in the spirit of Ref. [56] (but more general and the total energy has been correctly implemented). The biggest advantage of this mode is that the leading errors in the forces are corrected, and that the workfunction can be evaluated for asymetric slabs. The disadvantage is that the convergence to the electronic groundstate might slow down considerably (i.e. more electronic iterations might be required to obtain the required precision). It is recommended to use this mode only after pre-converging the wavefunctions without the LDIPOL flag, and the center of charge should be set by hand (DIPOL = center of mass). The user must also make sure that the cell is sufficiently large to determine the dipol moment with good accuracy. If the cell is too small, it is usually very difficult to tell whether charge is located on the ``left'' or ``right'' side of the slab, causing very slow convergence (often convergence improves with the size of the supercell).
Quadrupole corrections are only correct for cubic supercells
(this means that the calculated ![]() corrections are wrong for
charged supercells if the supercell is not cubic).
In addition we have found empirically that for charged systems
with excess electrons (NELECT
corrections are wrong for
charged supercells if the supercell is not cubic).
In addition we have found empirically that for charged systems
with excess electrons (NELECT![]() NELECT
NELECT
![]() ) more
reliable results can
be obtained if the energy after
correction of the linear error (
) more
reliable results can
be obtained if the energy after
correction of the linear error (![]() ) is plotted against
) is plotted against
![]() to extrapolate results manually for
to extrapolate results manually for ![]() . This
is due to the uncertainties in extracting the quadrupole moment
of systems with excess electrons.
. This
is due to the uncertainties in extracting the quadrupole moment
of systems with excess electrons.