Supported only by VASP.4.6 and on. THIS FEATURE IS IN LATE BETA STAGE (BUGS ARE POSSIBLE).
The L(S)DA often fails to describe systems with localized (strongly correlated)
![]() and
and ![]() electrons (this manifests itself primarily in the form of
unrealistic one-electron energies).
In some cases this can be remedied by introducing a strong intra-atomic interaction
in a (screened) Hartree-Fock like manner, as an on site replacement of the L(S)DA.
This approach is commonly known as the L(S)DA+U method.
electrons (this manifests itself primarily in the form of
unrealistic one-electron energies).
In some cases this can be remedied by introducing a strong intra-atomic interaction
in a (screened) Hartree-Fock like manner, as an on site replacement of the L(S)DA.
This approach is commonly known as the L(S)DA+U method.
VASP allows one to choose between two different approaches to L(S)DA+U:
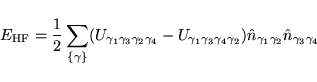
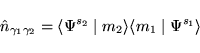
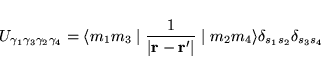
The unscreened e-e interaction
![]() can be written in terms of Slater's integrals
can be written in terms of Slater's integrals ![]() ,
, ![]() ,
, ![]() , and
, and ![]() (f-electrons).
Using values for the Slater integrals calculated from atomic wave functions, however,
would lead to a large overestimation of the true e-e interaction, since in solids
the Coulomb interaction is screened (especially
(f-electrons).
Using values for the Slater integrals calculated from atomic wave functions, however,
would lead to a large overestimation of the true e-e interaction, since in solids
the Coulomb interaction is screened (especially ![]() ).
).
In practice these integrals are therefore often treated as parameters,
i.e., adjusted to reach agreement with experiment in some sense: equilibrium volume, magnetic
moment, band gap, structure.
They are normally specified in terms of the effective on site Coulomb-
and exchange parameters, ![]() and
and ![]() .
(
.
(![]() and
and ![]() are sometimes extracted from constrained-LSDA calculations.)
are sometimes extracted from constrained-LSDA calculations.)
These translate into values for the Slater integrals in the following way (as implemented in VASP at the moment):
The essence of the L(S)DA+U method consists of the assumption that
one may now write the total energy as:
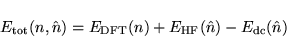
Currently VASP allows for the choice between two different definitions for the double counting energy:
| LSDA+U |
|
| LDA+U |
|
![\begin{displaymath}
E_{\rm LSDA+U}=E_{\rm LSDA}+\frac{(U-J)}{2}\sum_\sigma \left...
...n_{m_1,m_2}^{\sigma} \hat n_{m_2,m_1}^{\sigma} \right) \right]
\end{displaymath}](img303.gif)
This can be understood as adding a penalty functional to
the LSDA total energy expression that forces the on site occupancy
matrix in the direction of idempotency, i.e.,
![]() .
(Real matrices are only idempotent when their eigenvalues are either
1 or 0, which for an occupancy matrix translates to either fully
occupied or fully unoccupied levels.)
.
(Real matrices are only idempotent when their eigenvalues are either
1 or 0, which for an occupancy matrix translates to either fully
occupied or fully unoccupied levels.)
Note: in Dudarev's approach the parameters ![]() and
and ![]() do not enter seperately, only the difference
do not enter seperately, only the difference ![]() is
meaningfull.
is
meaningfull.
The L(S)DA+U in VASP is switched on by means of the following tags
NB: LDAUL, LDAUU, and LDAUJ must be specified for all
atomic species!
It is important to be aware of the fact that when using
the L(S)DA+U, in general the total energy will depend on the parameters
![]() and
and ![]() . It is therefore not meaningful to compare the total
energies resulting from calculations with different
. It is therefore not meaningful to compare the total
energies resulting from calculations with different ![]() and/or
and/or ![]() [c.q.
[c.q. ![]() in case of Dudarev's approach].
in case of Dudarev's approach].
Note on bandstructure calculation: The CHGCAR file also contains only information up to LMAXMIX for the on-site PAW occupancy matrices. When the CHGCAR file is read and kept fixed in the course of the calculations ( ICHARG=11), the results will be necessarily not identical to a selfconsistent run. The deviations can be (or actually are) large for L(S)DA+U calculations. For the calculation of band structures within the L(S)DA+U approach, it is hence strictly required to increase LMAXMIX to 4 (d elements) and 6 (f elements). (see Sec. 6.56).