The AE-part of the program rhfsps is controlled by the V_RHFIN file. This file is strictly formatted, and you must be very careful, if you change the file. Typically the file might have the following contents:
Pd : s1 d9, CA 11 46. .002000 106.42000 125. .50E-05 .100 200FCA 36.00000 .7 1.0 0 1.0 .0 .5-1761.5171 2.0000 2.0 .0 .5 -257.9015 2.0000 2.0 1.0 1.5 -231.7505 6.0000 3.0 .0 .5 -46.6977 2.0000 3.0 1.0 1.5 -38.0485 6.0000 3.0 2.0 2.5 -24.196610.0000 4.0 .0 .5 -6.4877 2.0000 4.0 1.0 1.5 -3.9976 6.0000 5.0 .0 .5 -.3403 1.0000 4.0 2.0 2.5 -.5091 9.0000 5.0 1.0 .5 -.1000 .0000The first line is a comment, which should contain the name of the element and the reference configuration for the valence electrons. The second line
11 46. .002000 106.42000 125. .50E-05 .100 200FCA 36.00000
J Z XION N AM H DELRVR PHI NC1 CH QCOR
|
GREEN
gives the most important information about the atom.
J is the number of orbitals, Z the ordering number.
XION can be used to supply a degree of ionization, but normally this value is zero.
N is the number
of grid points, usually we use 2000, AM the atomic mass, which is used
to calculate the innermost point for the logarithmic grid. H
determines the spacing between the grid points. The grid points are
given by
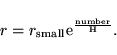 |
(51) |
| Slater-XC | |
| HL | Hedin Lundquist (1971) |
| CA | Ceperly and Alder parameterized by |
| J.Perdew and Zunger | |
| WI | Wigner interpolation |
| PB | Perdew -Becke |
| PW | Perdew -Wang 86 |
| LM | Langreth-Mehl-Hu |
| 91 | Perdew -Wang 91 |
Among these, the last four are gradient corrected functionals.
The parameter QCOR determines the number of core electrons (i.e.
non valence electrons).
The next line in the V_RHFIN file supplies less important
information. The first parameter is the SLATER parameter
used only in conjunction with the Slater-XC. The next parameter is no longer used,
and the last one can be used the set up so called latter correction
to the exchange correlation potential. Latter corrections must
not be applied if pseudopotentials are calculated. The remaining J lines
give information about each atomic orbital. The code is scalar relativistic,
but the inputfile is compatible to a relativistic input format.
The first value in each line is the main quantum number, the second one
the l-quantum number, and the third one the j-quantum number (![]() ).
The j-quantum number is not used in the program. The next value gives
the energy of the atomic orbital, the last number is the occupancy of the orbital.
The supplied energy is uncritical and only used as a start value
for the calculation of the atomic orbitals. As a starting guess you might insert
values obtained from an atom lying close to the atom of interest.
).
The j-quantum number is not used in the program. The next value gives
the energy of the atomic orbital, the last number is the occupancy of the orbital.
The supplied energy is uncritical and only used as a start value
for the calculation of the atomic orbitals. As a starting guess you might insert
values obtained from an atom lying close to the atom of interest.
The program rhfsps writes two files V_RHFOUT and V_TABOUT. The V_RHFOUT file is compatible to V_RHFIN and can be copied to V_RHFIN, if V_TABOUT is copied to V_TABIN. In this case rhfsps will start from the fully converged AE-potential supplied in V_TABIN. This saves time, and generally we recommend this setting.