


Next: Partial occupancies, different methods
Up: Theoretical Background
Previous: Wrap-around errors
Contents
Non-selfconsistent Harris-Foulkes functional
Recently there was an increased interest in the so called
Harris-Foulkes (HF) functional. This functional is non selfconsistent:
The potential is constructed for
some 'input' charge density, then the band-structure term is calculated for
this fixed non selfconsistent potential. Double counting corrections
are calculated from the input charge density: the functional
can be written as
![\begin{eqnarray*}\vspace*{1mm}
E_{\rm HF} [\rho_{\rm in},\rho] & = &
\mbox{ban...
...xc}_{\rm in}) \rho_{\rm in}] + E^{\rm xc}[\rho_{\rm in}+\rho_c].
\end{eqnarray*}](img470.gif)
It is interesting that the functional gives a good description of the
binding-energies, equilibrium lattice constants, and bulk-modulus even
for covalently bonded systems like Ge.
In a test calculation we have found that the pair-correlation function of l-Sb
calculated with the HF-function and the full Kohn-Sham functional
differs only slightly.
Nevertheless, we must point out that the computational gain in comparison to
a selfconsistent calculation is in many cases very small (for Sb less than 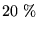 ).
The main reason why to use the HF functional is therefore to access and
establish the accuracy of the HF-functional, a topic which is currently
widely discussed within the community of solid state physicists. To our knowledge
VASP is one of the few pseudopotential codes,
which can access the validity of the HF-functional
at a very basic level, i.e. without any additional restrictions like local
basis-sets etc.
).
The main reason why to use the HF functional is therefore to access and
establish the accuracy of the HF-functional, a topic which is currently
widely discussed within the community of solid state physicists. To our knowledge
VASP is one of the few pseudopotential codes,
which can access the validity of the HF-functional
at a very basic level, i.e. without any additional restrictions like local
basis-sets etc.
Within VASP the band-structure energy is exactly evaluated using
the same plane-wave basis-set and the same accuracy which is used for
the selfconsistent calculation. The forces and the stress tensor are correct,
insofar as they are an exact derivative of the Harris-Foulkes functional.
During a MD or an ionic relaxation the charge density is correctly updated at each
ionic step.
Next: Partial occupancies, different methods
Up: Theoretical Background
Previous: Wrap-around errors
Contents
Georg Kresse
2009-04-23