The following section discusses the minimization algorithms implemented in VASP. We generally have one outer loop in which the charge density is optimized, and one inner loop in which the wavefunctions are optimized. Have at least a look at the flowchart.
Most of the algorithms implemented in VASP use an iterative matrix-diagonalization scheme: the used algorithms are based on the conjugate gradient scheme [20,21], block Davidson scheme [22,23], or a residual minimization scheme - direct inversion in the iterative subspace (RMM-DIIS) [19,26]). For the mixing of the charge density an efficient Broyden/Pulay mixing scheme[24,25,26] is used. Fig. 3 shows a typical flow-chart of VASP. Input charge density and wavefunctions are independent quantities (at start-up these quantities are set according to INIWAV and ICHARG). Within each selfconsistency loop the charge density is used to set up the Hamiltonian, then the wavefunctions are optimized iteratively so that they get closer to the exact wavefunctions of this Hamiltonian. From the optimized wavefunctions a new charge density is calculated, which is then mixed with the old input-charge density. A brief flow chart is given in Fig. 3.
1cm (0,0)[bc] (0,0.5) (-6,-1)3.0 (0,0.5) ( 6,-1)3.0 (0,-.5) (-6, 1)3.0 (0,-.5) ( 6, 1)3.0 (-3,0.1) (0,0)[br]no
(13.0,1.0)[bl] (0,0.0) ( 1, 6)0.166 (0,0.0) ( 1, 0)12.8 (12.8,0.0) ( 1, 6)0.166 (.166,1.0) ( 1, 0)12.8
The conjugate gradient and the residual minimization scheme do not recalculate the exact Kohn-Sham eigenfunctions but an arbitrary linear combination of the NBANDS lowest eigenfunctions. Therefore it is in addition necessary to diagonalize the Hamiltonian in the subspace spanned by the trial-wavefunctions, and to transform the wavefunctions accordingly (i.e. perform a unitary transformation of the wavefunctions, so that the Hamiltonian is diagonal in the subspace spanned by transformed wavefunctions). This step is usually called sub-space diagonalization (although a more appropriate name would be, using the Rayleigh Ritz variational scheme in a sub space spanned by the wavefunctions):
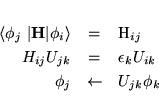
In general all iterative algorithms work very similar:
The core quantity is the residual vector
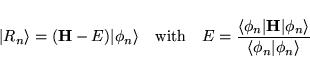 |
(44) |
